Title
Page MR
Model Brookhaven Structures
Journal
References
Final Thought
Jump
To: Binding Domain
Ligand
Binding
The nuclear receptors constitute a super family of protein hormones that serve as transcription factors. They typically reside in the cytosol and, after ligand binding, migrate to the nucleus to exert their biological action. The ligands are lipophilic small molecules including retinoids, steroids, thyroxine, and vitamin D. These receptors are powerful regulators of developmental processes including cellular division and differentiation, normal homeostasis and the stress response. An understanding of their structure might provide clues for manipulation of the ligand-protein interactions involved in their function with the ultimate goal of designing drugs to mediate hormonal control of body processes. The magnitude of current research on these proteins is tracked by several "resource" sites: Androgen Receptors , Glucocorticoid Receptors, Peroxisome Proliferator Receptors, and a site devoted to Proteins Associated With Steroid Receptors.
The nuclear receptors have approximately 400-900 amino acid
residues that are divided into three domains: the N-terminal domain whose
function is still unclear, the highly conserved DNA binding domain and the
C-terminal ligand binding region. A
sequence alignment of known nuclear receptors is available at the PFam
site. A schematic of the nuclear receptor sequence is shown below in
figure 1.
Figure 1: Schematic Of Protein Sequence For Nuclear Receptors
This site will focus on the ligand-binding domain (LBD) at the C terminal region of these receptors.
Nuclear Receptors |
Ligand Binding
The ligand binding domain of the nuclear receptors is
classified by SCOP
as an all-alpha protein. The CATH
site ( search for CATH code 1.10.565.10 or nuclear receptor) provides a summary of the topological properties for
this group The crystal structures for a number of these nuclear proteins from
the Brookhaven Data Base have been
deposited here The first crystal
structure of this family to be determined was a heterodimer of Retinoic
Acid . The monomer
of retinoic acid (with water of
crystallization and bound ligand) shows the tri-fold arrangement of helix
cylinders that is characteristic of this nuclear super family. The receptors are
commonly illustrated with the predominantly hydrophobic ligand binding cavity on
the bottom of the image. An annotated image of the ligand binding domain of the
retinoic acid receptor is shown below in figure 2
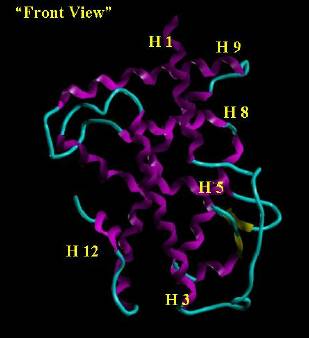
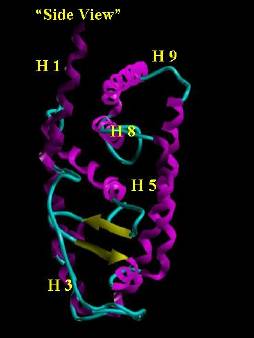
Figure 2. Orthographic View (image on the right is the
image on the left rotated 90 degrees clockwise
on the y axis) of the Retinoic Acid Receptor. Helix regions are colored magenta
and beta sheets are yellow. The cyan tube traces the backbone between regions of
defined secondary structure. The ligand binding domain is the cavity on the
"bottom" of the receptor in the volume defined by H 3, H 5, H 12 and the
"loop" (the yellow beta sheets including the cyan hairpin turn).
Although the general topology of the receptor fold is common among the receptor types, there is variation in the length and composition of the helical segments and the loop region. The arrangement of helical segments around this "lower-central" cavity gives rise to the canonical tri-fold (three distinct layers) of this super family. Looking at the "side-view" in figure 2, the three helical fold domains can be visualized as "lower left (around H3)," "central, skewed left (around H 5) " and the “right-side" (around" H 8 & 9). This segregation into partitions suggests a conformational mobility of the receptor upon ligand binding. It may also represent separate volumes brought together by the actions of chaperones (proteins that assist in the folding of proteins). This is supported by the observation that protease digestions of bound and unbound ligands give different profiles. The strongest evidence for conformational change comes from crystallographic structures of ligand bound and unbound nuclear receptors showing different locations for H 12. The working hypothesis is that in the unbound receptor, H 12 lies "below" the receptor exposing the central binding cavity. This makes the interior region of the receptor accessible to the approaching ligand.
The abundance of hydrophobicity lining the ligand binding cavity of the nuclear receptors is shown below by the predominance of green (hydrophobic) residues in figure 3.
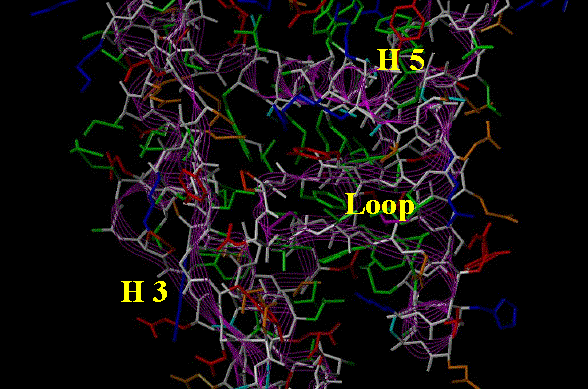
Figure 3. Stick
representation of the binding pocket of a nuclear receptor. Residues are colored
as follows: Acidic (Red), Basic (Blue), Hydrophobic (Green), and Neutral-Polar
(Orange). The backbone (White) is traced with a Magenta ribbon. The ligand
binding domain is the open volume bounded by H3, H 5 and the Loop. The potential
H bonding residues (orange, blue and red) that are adjacent to this volume are
primarily at either end of this space with the central region clearly
hydrophobic in nature.
Nuclear Receptors | Ligand Binding
A steroid can be envisioned as a predominantly hydrophobic moiety with polar projections. An example, aldosterone hemi-acetal, is shown below in figures 4 & 5.
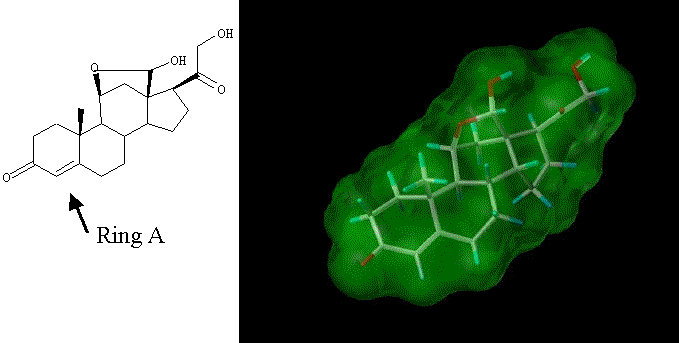
Figure 4. Aldosterone Hemi-Acetal
Figure 5. Stick representation of aldosterone hemi-acetal surrounded by its Connolly surface contour showing a hydrophobic core with polar projections. Colors: Carbon (White), Hydrogen (Cyan), Oxygen (Red) and the Connolly Contour (Green)
With H 12 "below" the receptor, the binding cavity is exposed to the exterior. Entropy (hydrophobic ligand seeks to avoid polar aqueous surroundings) would favor approach of the ligand into the receptor cavity. One polar end (unsaturated carbonyl on steroid ring A) is directed towards an Arg, Gln, or Asn (depends on receptor) in the volume near the hairpin turn to "anchor" the ligand to the receptor. Once the ligand binding domain cavity is occupied by the ligand, H 12, by hydrogen bonding to the other polar end of the steroid "closes" the cavity. This most likely would result in conformational shifts of the helix regions bounded by the "incoming" H 12. Thus, this H 12 ligand-contact region could play a role in determining ligand selectivity. Movement of adjacent helix regions could redefine surface contact areas with associated proteins and other molecules present within the cell. This sequence of events is represented schematically in the three figures below.
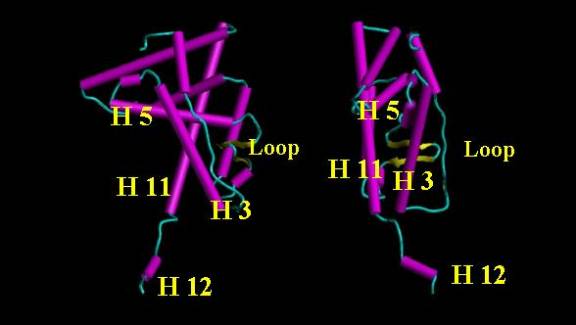
Figure 6. Orthographic view of a nuclear receptor with H 12 below. The volume defined by H 12, H 11, H 5 and H 3 provides an opening into the ligand binding domain region. Polar regions on H 5 and H 3 near the "loop" assist in docking the polar regions (eg. carbonyl on the A ring of a steroid) of the ligand.
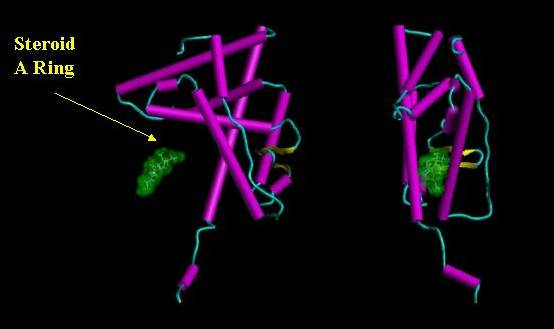
Figure 7. Orthographic view of a steroid (green surface contour) approaching the ligand binding cavity.
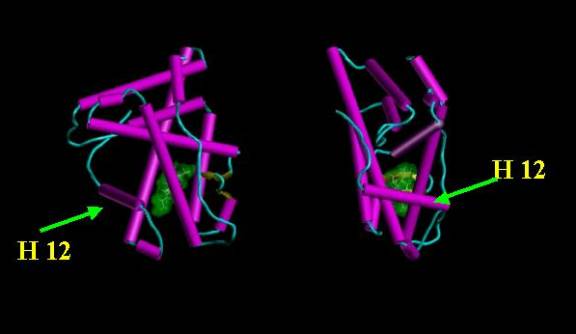
Figure 8. Orthographic view of receptor after H 12 has closed the ligand binding cavity.
This results in the predominantly lipophilic ligand being surrounded by the hydrophobic interior of the receptor.
Since H 12 is relatively mobile, its final resting position with respect to H 11 and H 3 will be sequence and ligand dependent. Changes in nature of residues in this area and the length of the sequence between H 11 and H 12 will influence the final "resting" location of H 12. This makes this region a prime focal area for ligand structure-activity studies.
Jump
To: Nuclear
Receptors | Binding
Domain | Ligand
Binding
Title
Page MR
Model Brookhaven Structures
Journal
References
Final Thought