Introduction
The field of catalysis is driven forward by the development of new paradigms for rapid, selective transformations of new and existing feedstocks into valuable products. Underpinning these developments are fundamental descriptors of mechanism, the atomic and electronic details that govern chemical reactivity. Quantum chemical simulation provides direct access at this level of information, but such simulation often comes with surprisingly high costs in terms of computational time and human effort -- costs that our research is working to eliminate.
New Methods for Automated Mechanism Searching
Research in the Zimmerman group seeks to revolutionize the characterization of mechanism using automated methods that are fast, reliable, and require minimal human effort. Techniques such as fast string methods for locating reaction paths can be combined with automatic elementary step exploration to yield quantitative comparisons of competing reaction paths. The methods can be operated as “single-ended,” where reactivity is explored without knowing the final destination, or “double-ended,” where the product is already known beforehand.
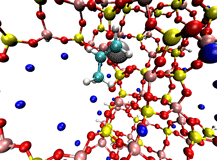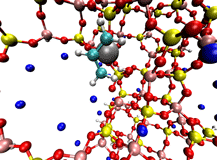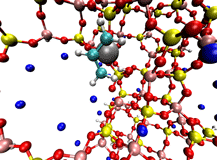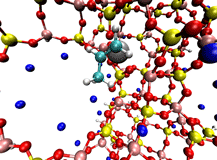 On the right is an example double-ended reaction path for Ni zeolite X catalyst activation involving propene. This reaction changes the inactive form of the catalyst into a form that undergoes highly selective olefin dimerization. New tools, such as enhancements to the Growing String Method to locate exact transition states and reaction paths simultaneously, are being created to more rapidly and reliably find the chemically-relevant pathways connecting important intermediates in catalytic systems.
On the right is an example double-ended reaction path for Ni zeolite X catalyst activation involving propene. This reaction changes the inactive form of the catalyst into a form that undergoes highly selective olefin dimerization. New tools, such as enhancements to the Growing String Method to locate exact transition states and reaction paths simultaneously, are being created to more rapidly and reliably find the chemically-relevant pathways connecting important intermediates in catalytic systems.
In a single-ended reactivity search, systematic automated investigation of pathways will locate reaction paths that would not have been found by manual investigation, and this means the predictive value can be high. Much of the “human bias” factor is removed from the simulations, leading to possibilities that have never been before conceived. Significant amounts of effort are currently being applied to creation of these tools, with the first algorithm being detailed in reference 1 below.
New Methods for Catalyst Optimization
When a catalyst operating mechanism is reasonably well-understood, the limiting factors for catalysis can be targeted for enhancement. New techniques are under development which will optimize specific catalyst properties using quantum chemical simulation. Stay tuned for more information as these exciting new opportunities for catalyst design are created and tested on challenging problems in renewable energy and polymerization catalysis.
Recent catalysis related publications:
1. P. M. Zimmerman, “Automated discovery of chemically reasonable elementary reaction steps,” in press, Journal of Computational Chemistry, (2013).
2. P. M. Zimmerman, “Navigating Molecular Space for Reaction Mechanisms: An Efficient, Automated Procedures,” accepted, Molecular Simulation (2014).
3. P. M. Zimmerman, “Growing string method with interpolation and optimization in internal coordinates: Method and examples,” Journal of Chemical Physics, 138, 184102 (2013).
4. P. M. Zimmerman, D. Tranca, J. Gomes, D. Lambrecht, A. T. Bell, M. Head-Gordon, “Ab Initio Simulations Reveal that Reaction Dynamics Strongly Affect Product Selectivity for the Cracking of Alkanes over H-MFI,” Journal of the American Chemical Society, 134, 19468-19476 (2012).
5. A. Mlinar, P. M. Zimmerman, F. E. Celik, M. Head-Gordon, A. T. Bell, “Effects of Brønsted Acid Site Proximity on the Oligomerization of Propene in H-MFI,” Journal of Catalysis, 288, 65-73 (2012).
6. P. M. Zimmerman, M. Head-Gordon, A. T. Bell, “Selection and validation of charge and Lennard-Jones parameters for QM/MM simulations of hydrocarbon interactions with zeolites,” Journal of Chemical Theory and Computation, 7(6), 1695-1703 (2011).