Links: this page
Early studies
Zap-70
T cell synapses
Correction of age effects
Work in progress
Research index
Miller Lab home page
1. Earlier studies.
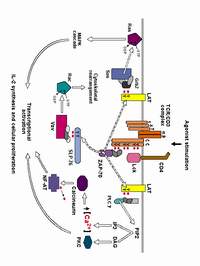
A series of studies over the last 15 years has shown that T cells from aged mice show multiple defects in the first few minutes of the activation process triggered by mitogens or by antibodies to T cell receptors. Defective proliferation and cytokine production (detectable 12 - 48 hr after activation) reflects diminished activation of immediate early genes, such as myc (detectable 2 - 4 hr after activation), which in turn depends upon a failure to generate second messengers at even earlier time points. T cells from old mice show declines in multiple aspects of the signal transduction process, all detectable within 2 - 15 minutes of activation, including:
- blunted rise in intracellular free calcium ion concentration
- roughly two-fold decline in activation of the cascade by which Raf-1 phosphorylates MEK, which in turn phosphorylates ERK (MAPK), which in turn phosphorylates a wide range of substrates including RS6 kinase
- decline in activation of JNK, a key mediator of the commitment to activation and proliferation. Curiously, JNK activation by a variety of forms of cellular stress is not altered by aging in mice.
- failure to phosphorylate the ITAM regions of the CD3z chain
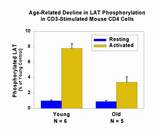
- failure to phosphorylate LAT, a key event in the formation of multi-component signal transduction complexes at the plasma membrane
Association of the protein kinase Zap-70 with the phosphorylated ITAM regions of CD3z is thought to be a critical stage for T cell activation, allowing Zap-70 access to its own substrates, including LAT. The decline with age in ITAM phosphorylation leads to the expectation of that Zap-70 association with the T cell receptor (TCR) should decline with age, leading in turn to the observed decline in LAT phosphorylation. It was thus a surprise to note an age-associated increase in the amount of Zap-70 associated with the CD3 complex in T cells of aged mice. How can Zap-70 associate with the TCR in old mice, if the TCR is hypo-phosphorylated? Why is LAT phosphorylation lower if there is a high level of TCR-associated Zap-70? This puzzling set of observations was further complicated by the observation that T cells from old and young mice had equivalent levels of Zap-70 enzyme function a few minutes after activation. If Zap-70 was turned on to an equal extent in young and old T cells, then why was there a defect in LAT phosphorylation? [Back to top]
3. Defects in T cell synapse formation.
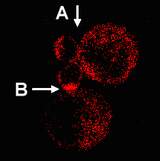
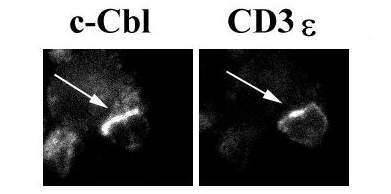
Confocal microscopic methods that monitor the location of protein kinases and their substrates within individual cells provided new insights into the molecular basis for T cell activation defects in aged mice. These studies showed that aging leads to a decline in the proportion of T cells in which activation induces translocation of kinases and substrates into the "synapse" area at which the T cell is in contact with its antigen-presenting cell (APC). T cells respond to APC stimulation by forming a "supermolecular activation complex," or synapse, containing the CD3/TCR complex, kinases including Zap-70 and protein kinase Cq, and substrates including LAT, Vav, and many others. The close proximity of kinases, substrates, and adaptor molecules in this synapse triggers a series of downstream events leading eventually to cytokine production and clonal proliferation.
The data show that about 50% of CD4 T cells from young mice will respond to an APC by synapse formation, but only about 25% of T cells from old mice can do so. Experiments in which different components of the synapse were labelled with contrasting fluorescent dyes established that synapse formation is essentially an "all or nothing" phenomenon, in that T cells that fail to translocate any of the component elements to the synapse fail to show translocation of any of these proteins. Studies of the response of T cells from a TCR-transgenic mouse line showed that the aging defect applied to naive T cells and to peptide antigens as well as to responses to stronger, polyclonal activators like antibodies to the CD3 complex. Studies of PKCq translocation showed that both CD4 and CD8 cells were affected by aging. Studies using fluorescent methods to identify specific T cell subsets showed that synapse formation was especially defective in a T cell subset (i.e. cells with high level P-glycoprotein expression) already shown in functional studies to be common in old mice and anergic in mice of any age.
These observations explain a part of the Zap-70 paradox described above: phosphorylation declines with age in T cells not because of a failure to activation Zap-70, but because of a failure to move kinase substrates, such as LAT and Vav, into the synapse region where they could come into contact with activated kinases. [Back to top]
4. T cell cytoskeletal abnormalities
Further work using T cell receptor transgenic mice, which respond only to a specific peptide, provided additional insight into the molecular basis for signal transduction defects in T lymphocytes from aged mice. The key experiment was the demonstration that transgenic T cells from old mice fail to respond even to antagonist peptides, i.e. to peptides that differ from the correct peptide at a single amino acid residue. These antagonist peptides do not trigger full synapse formation, but they do trigger (in young T cells) initial steps towards reorganization of the cytoskeleton. The failure of aged T cells to initiate cytoskeletal changes shows that their defect lies at a stage prior to recognition, by the T cell receptor, of the specific residues that distinguish agonist from antagonist peptides. Further studies showed an age-related defect in the ability of T cells to link T cell receptor molecules to the cytoskeletal matrix [PubMed]. More recent work [PubMed] has focused attention on molecules in the ezrin/moesin/radixin (ERM) family, and documented age-related defects in ERM phosphorylation, associations with key surface proteins such as CD43 and CD44. T cells from aged mice also show defects in two of the G-proteins known to control ERM function. [Back to top]
5. Correction of age-related defects in synapse formation
Our most recent work on T cell activation has suggested both a new molecular model for T cell senescence and an approach to reversal of age-related T cell immune defects. Studies of the surface protein CD43 suggested that T cells from aged mice had increased levels of heavily glycosylated proteins. Remarkably, the heavily glycosylated molecules were found principally in a specific subset of CD4 T cells that had been shown in previous work to be anergic and to accumulate in older mice. These findings led to a model in which heavily glycosylated surface molecules, unable to interact with cytoskeletal motors and thus unable to leave the synapse, interfered with proper T cell receptor binding to antigen-bearing stimulator cells.
A key test of the model was the demonstration that enzymatic removal of these glycoproteins could restore T cell functions. Treatment with OSGE (O-sialoglycoprotein endoprotease), a bacterial enzyme, was indeed found able to restore synapse formation CD4 and CD8 T cells of aged mice [PubMed]. OSGE treatment can also restore to aged T cells the ability to produce IL-2 and IL-4, express activation antigens such as CD69 and the IL-2 receptor CD25, and generate cytotoxic effector cells. OSGE affects even very early stages in the activation process, such as the increased internal calcium ion concentration seen within the first 3 minutes after stimulation [PubMed]. OSGE can also augment calcium signals and cytokine production in T cells of young mice, suggesting that some OSGE-sensitive targets interfere with T cell stimulation in young adults as well. The target molecules that mediate the OSGE effects are not known, but the effect of the enzyme on CD43 null mice suggests that other proteins carrying O-linked sialylated glycans, such as CD44 and CD45, may be important [PubMed]. Back to top]
Work is in progress on several fronts to gain additional insight into T cell activation defects. These studies include:
- Biochemical analyses to determine which glycoproteins are involved in the ability of OSGE and other enzymes to improve function of aged T cells.
- Further studies of cytoskeletal abnormalities to evaluate upstream signaling through G protein activation and downstream consequences on synapse formation and gene activation.
- Adoptive transfer studies to see if OSGE treatment of T cells can improve their ability to protect mice from viruses and neoplasia.
Researchers: Gonzalo Garcia, Amir Sadighi-Akha
Alumni: Ami Tamir, Chris Kirk, Mike Eisenbraun, Scott Berger
Support: NIA grant AG019619
[last update: December, 2007]
Signal Pathways in T Cells
LAT Phosphorylation Defect
PKC-q
at Synapse
c-Cbl and CD3e
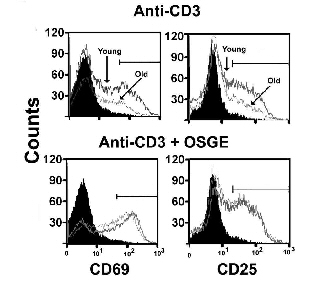
OSGE repairs CD69 and CD25 activation in aged CD4 cells