 |
Johann Gagnon-BartschAssociate Professor Contact InfoE-mail: johanngb@umich.edu Office: 441 West Hall Phone: (734) 763-1427 Mailing address: |
| |
Johann Gagnon-BartschAssociate Professor Contact InfoE-mail: johanngb@umich.edu Office: 441 West Hall Phone: (734) 763-1427 Mailing address: |
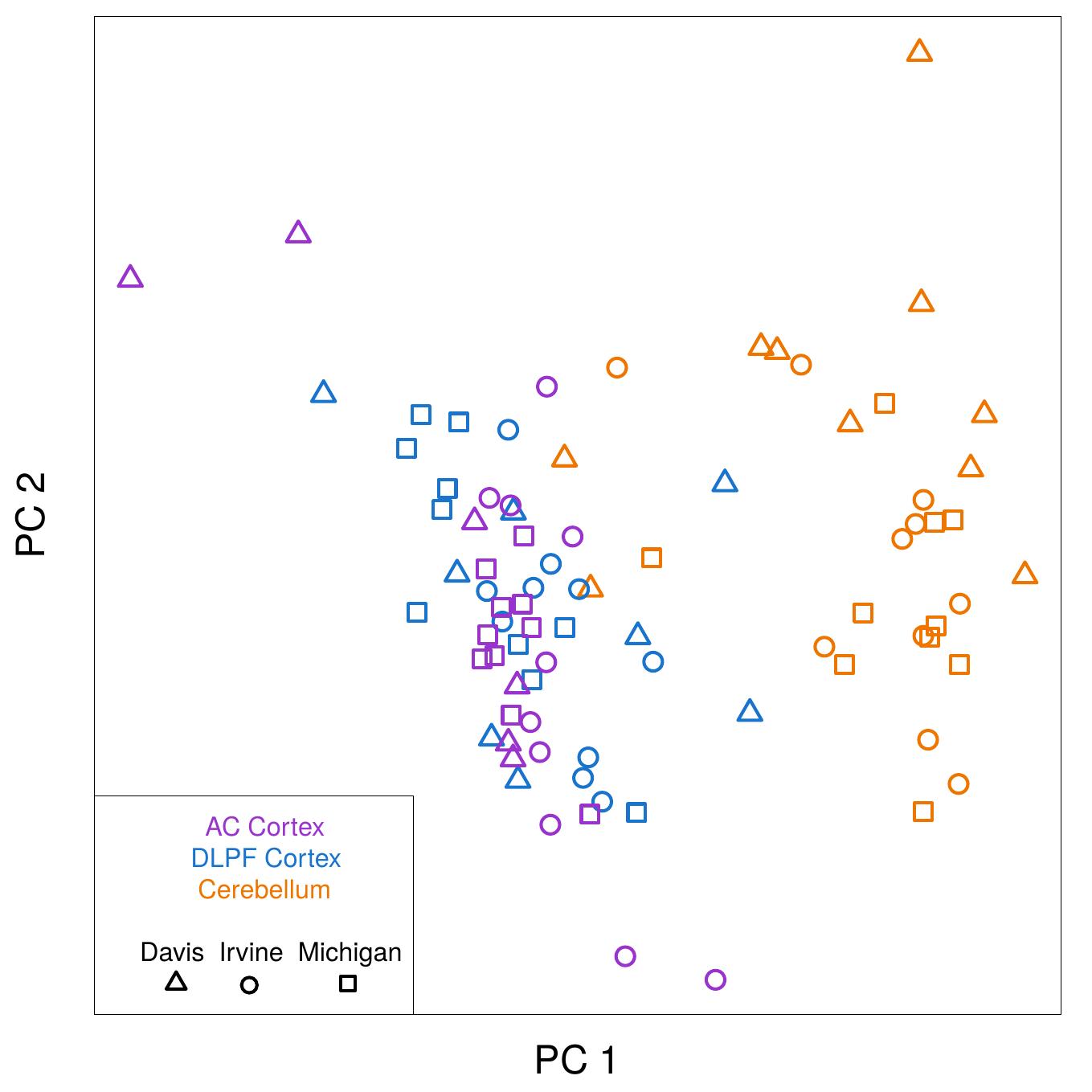 RUV is a set of methods originally developed to remove batch effects and other unwanted variation from gene expression data. More generally, RUV attempts to adjust high dimensional data for unobserved confounders, by making use of negative controls and replicates. A negative control is a variable that is known a priori to be (1) unaffected by the factor of interest, and (2) affected by the unobserved confounders. Negative controls and replicates can be used to help identify unwanted variation and separate it from variation of interest, even when the wanted and unwanted variation are correlated, and even when the factors causing the unwanted variation are unknown.
RUV is a set of methods originally developed to remove batch effects and other unwanted variation from gene expression data. More generally, RUV attempts to adjust high dimensional data for unobserved confounders, by making use of negative controls and replicates. A negative control is a variable that is known a priori to be (1) unaffected by the factor of interest, and (2) affected by the unobserved confounders. Negative controls and replicates can be used to help identify unwanted variation and separate it from variation of interest, even when the wanted and unwanted variation are correlated, and even when the factors causing the unwanted variation are unknown.
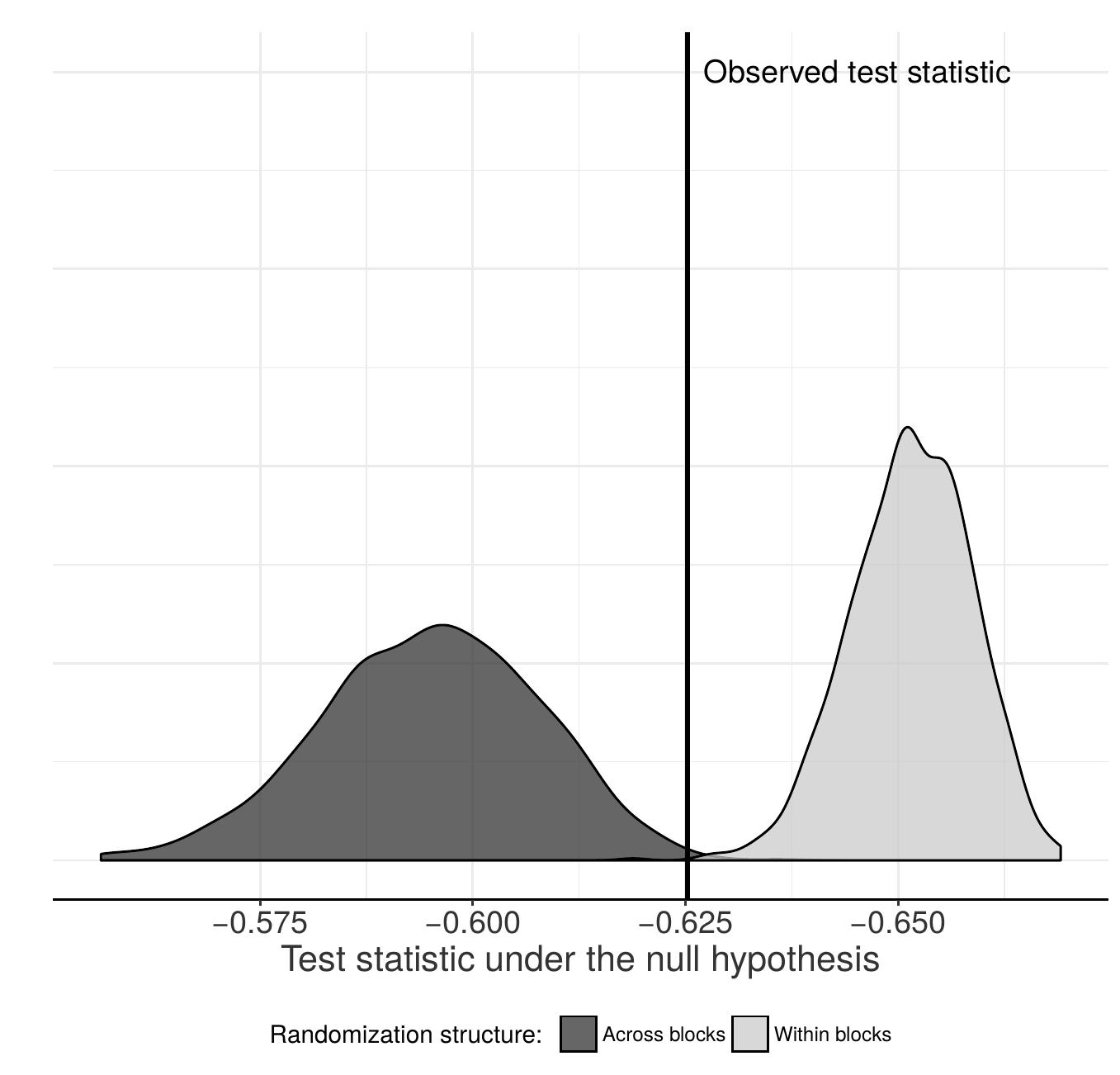 In many studies in the social sciences and medicine the researcher does not control treatment assignment and instead may rely upon natural experiments or matching methods as a substitute to experimental randomization. In such cases it is helpful to check whether observed covariates are balanced across treatment conditions. The Classification Permutation Test (CPT) is a covariate balance test that first trains a classifier to distinguish treated units from control units, and then, using permutation inference, determines whether the classifier is able to do so better than would be expected by chance.
In many studies in the social sciences and medicine the researcher does not control treatment assignment and instead may rely upon natural experiments or matching methods as a substitute to experimental randomization. In such cases it is helpful to check whether observed covariates are balanced across treatment conditions. The Classification Permutation Test (CPT) is a covariate balance test that first trains a classifier to distinguish treated units from control units, and then, using permutation inference, determines whether the classifier is able to do so better than would be expected by chance.
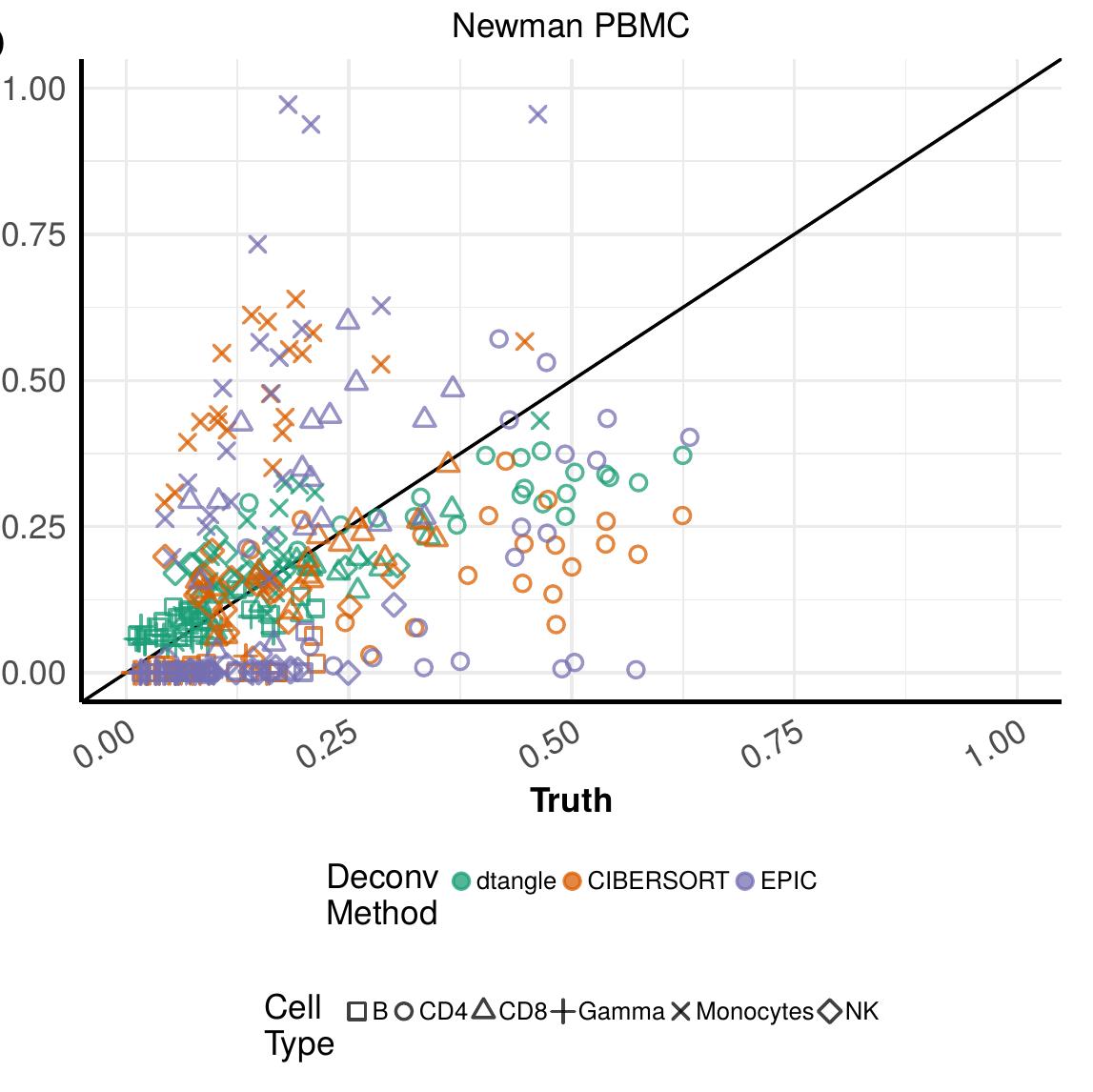 Biological tissues are typically composed of several distinct cell types. dtangle is a method to estimate the proportions of different cell types comprising a tissue sample from gene expression data. (This is sometimes referred to as "cell type deconvolution.") Similar to other deconvolution methods, dtangle requires reference expression profiles for each cell type, as well as a list of marker genes that are expressed primarily in one cell type. Where dtangle is unique is in its treatment of scale; gene expression values are considered on both linear and log scales, with the dual aims of a scientifically plausible mixing model, and statistical robustness of the fitting procedure.
Biological tissues are typically composed of several distinct cell types. dtangle is a method to estimate the proportions of different cell types comprising a tissue sample from gene expression data. (This is sometimes referred to as "cell type deconvolution.") Similar to other deconvolution methods, dtangle requires reference expression profiles for each cell type, as well as a list of marker genes that are expressed primarily in one cell type. Where dtangle is unique is in its treatment of scale; gene expression values are considered on both linear and log scales, with the dual aims of a scientifically plausible mixing model, and statistical robustness of the fitting procedure.
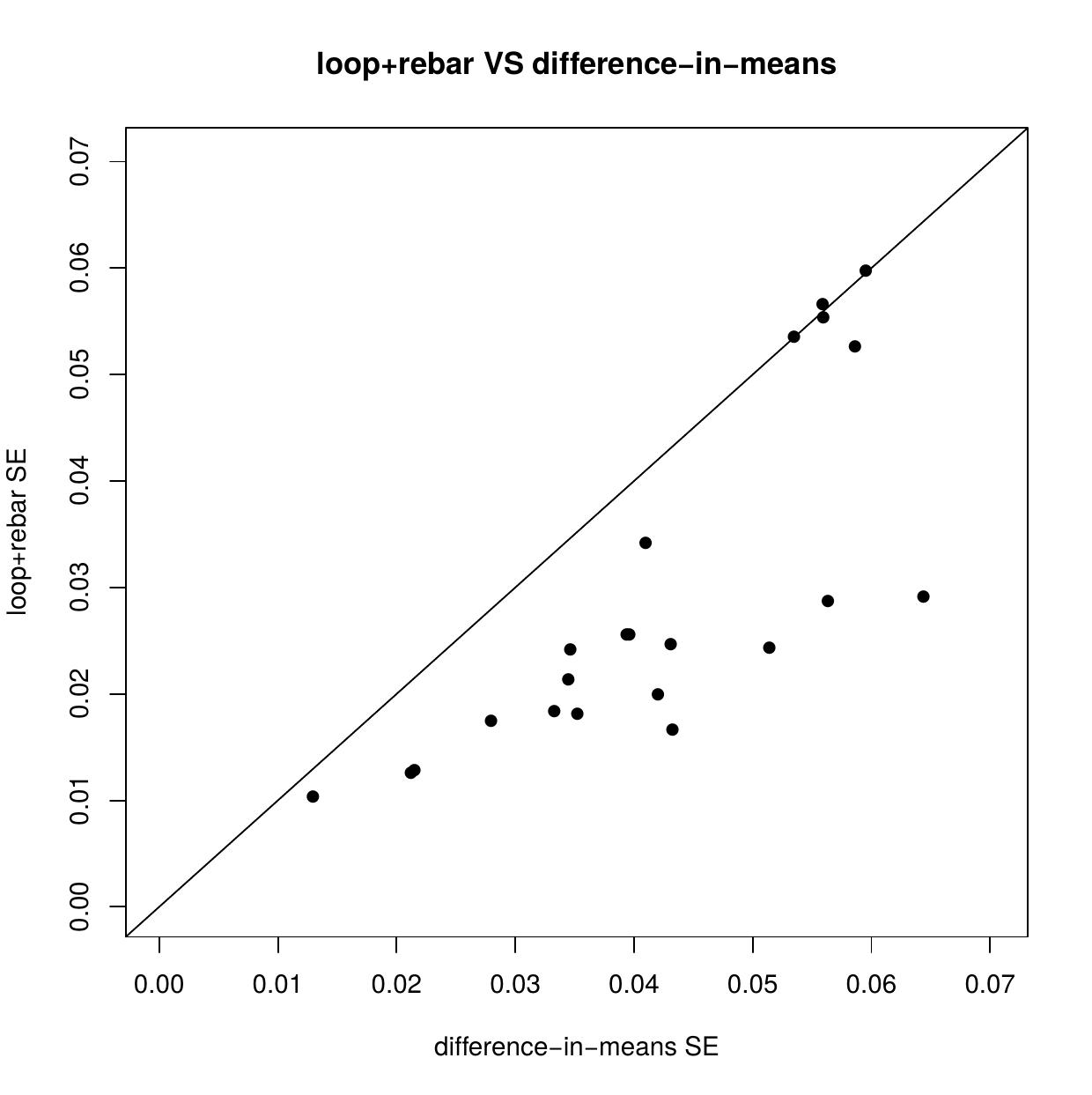 Two advantages of randomized trials are (1) potential confounding variables are largely balanced across treatment conditions, and (2) design-based inference may be used, in which statistical assumptions are largely justified by the physical act of randomization. Randomization does not balance potential confounders perfectly, however, and there are typically small observed imbalances in baseline covariates. Adjusting for these imbalances can improve the precision of treatment effect estimates, but methods that do so are not always design-based. We are working on developing new design-based estimators to fill this gap.
Two advantages of randomized trials are (1) potential confounding variables are largely balanced across treatment conditions, and (2) design-based inference may be used, in which statistical assumptions are largely justified by the physical act of randomization. Randomization does not balance potential confounders perfectly, however, and there are typically small observed imbalances in baseline covariates. Adjusting for these imbalances can improve the precision of treatment effect estimates, but methods that do so are not always design-based. We are working on developing new design-based estimators to fill this gap.
 The immediate physical and bio-chemical surroundings of a cell, the cellular microenvironment, is an important component of many fundamental cell and tissue level processes and is implicated in many diseases and dysfunctions. To study perturbations of cellular microenvironments a novel image-based cell-profiling technology called the microenvironment microarray (MEMA) has been recently employed. We are helping to develop an analysis pipeline for MEMA data, which will allow for the integration of image features, as well as adjustments for spatial and other technical artifacts in the data.
The immediate physical and bio-chemical surroundings of a cell, the cellular microenvironment, is an important component of many fundamental cell and tissue level processes and is implicated in many diseases and dysfunctions. To study perturbations of cellular microenvironments a novel image-based cell-profiling technology called the microenvironment microarray (MEMA) has been recently employed. We are helping to develop an analysis pipeline for MEMA data, which will allow for the integration of image features, as well as adjustments for spatial and other technical artifacts in the data.
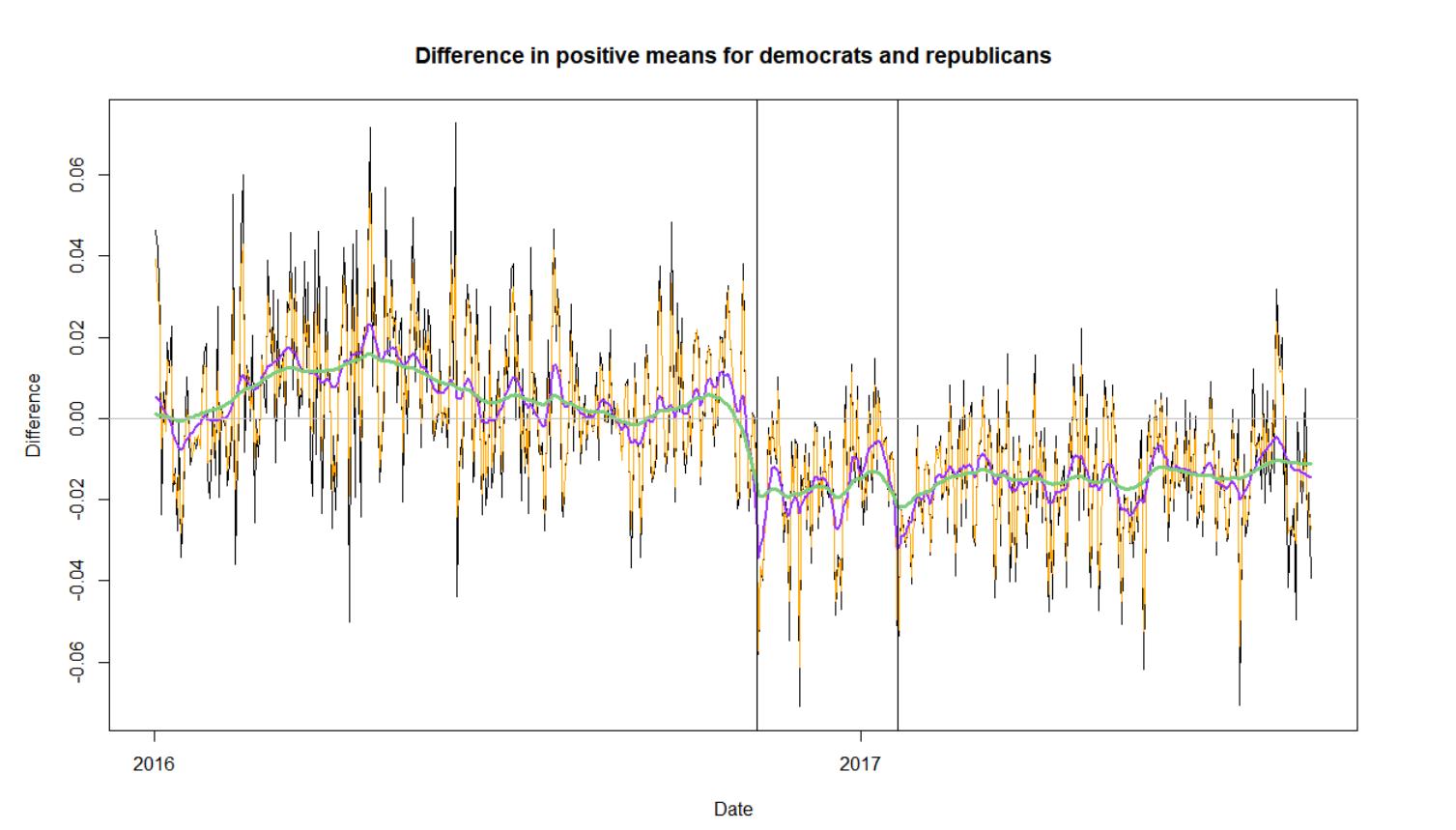 Traditional surveys, such as those used for political polling and economic research, are expensive and suffer increasingly from non-response. Social media has been suggested as an alternative data source to augment or even replace survey data. We are exploring the feasibility of using social media data in this manner.
Traditional surveys, such as those used for political polling and economic research, are expensive and suffer increasingly from non-response. Social media has been suggested as an alternative data source to augment or even replace survey data. We are exploring the feasibility of using social media data in this manner.
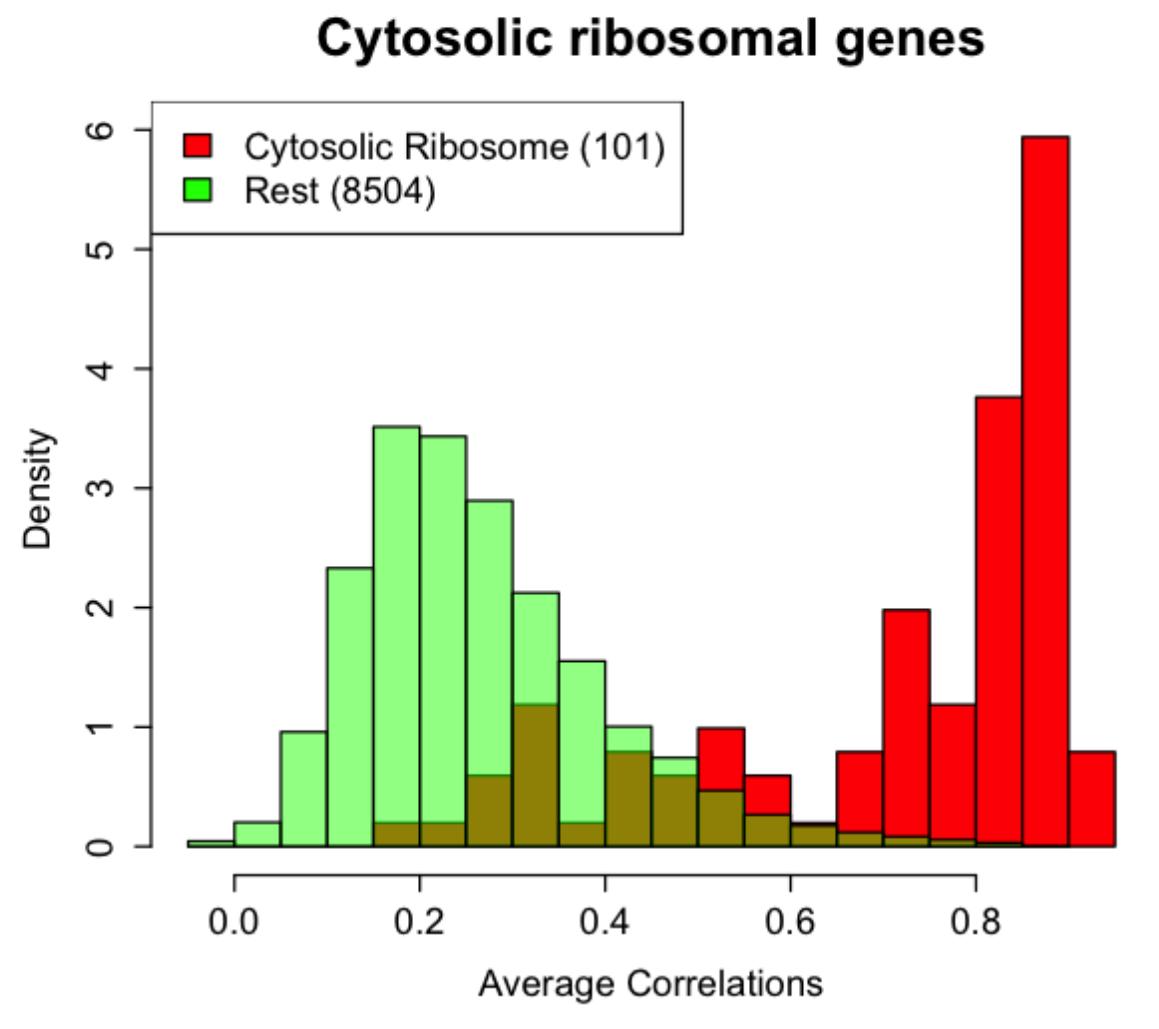 Genes with relatively stable expression are of biological interest, and also useful for normalization (see RUV above). We are interested in discovering such genes, and especially interested in genes that are stable even at the level of single cells. At the single cell level, the notion of stability is somewhat ambiguous; for example, a gene could be stable in terms of the absolute quantity of transcripts, or in terms of concentration (proportional to cell size). Our goal is to identify different sets of genes that satisfy different notions of stability.
Genes with relatively stable expression are of biological interest, and also useful for normalization (see RUV above). We are interested in discovering such genes, and especially interested in genes that are stable even at the level of single cells. At the single cell level, the notion of stability is somewhat ambiguous; for example, a gene could be stable in terms of the absolute quantity of transcripts, or in terms of concentration (proportional to cell size). Our goal is to identify different sets of genes that satisfy different notions of stability.